| February 28, 2003 |
|
 |
 |
| Scientists Find That Apes and Monkeys Provide Needed Help in Understanding the Human Genome | |
|
Contact: Lynn Yarris (510) 486-5375 lcyarris@lbl.gov |
|
|
|
BERKELEY, CA � Scientists with the U.S. Department of Energy's Joint Genome Institute (JGI) and the Lawrence Berkeley National Laboratory (Berkeley Lab) have developed a powerful new technique for deciphering biological information encoded in the human genome. Called "phylogenetic shadowing," this technique enables scientists to make meaningful comparisons between DNA sequences in the human genome and sequences in the genomes of apes, monkeys, and other non-human primates. With phylogenetic shadowing, scientists can now study biological traits that are unique to members of the primate family.
"Now that the sequence of the human genome has almost been completed the next challenge will be the development of a vocabulary to read and interpret that sequence," says Edward Rubin, M.D., director of the Joint Genome Institute (JGI) for the U.S. Department of Energy, and Berkeley Lab's Genomics Division, who led the development of the phylogenetic shadowing technique. "The ability to compare DNA sequences in the human genome to sequences in non-human primates will enable us in some ways to better understand ourselves than the study of evolutionarily far-distant relatives such as the mouse or the rat," Rubin adds. "This is important because as valuable as models like the mouse have been, there are many physical and biochemical attributes of humans that only other primates share." Using phylogenetic shadowing, Rubin and his colleagues were able to identify the DNA sequences that regulate the activation or "expression" of a gene that is an important indicator of the risk for heart disease and is found only in primates. The results of this research are reported in a paper published the February 28 issues of the journal Science. Co-authoring the paper with Rubin were Dario Boffelli, Dmitriy Ovcharenko, Keith Lewis and Ivan Ovcharenko of Berkeley Lab, plus Jon McAuliffe and Lior Pachter, of the University of California at Berkeley. Comparative genomics, comparing segments of DNA in the human genome to DNA segments in the genomes of other organisms that have been sequenced, such as the mouse, the puffer fish or the sea squirt, has proven to be an effective means of identifying genes, the DNA sequences that code for proteins, and gene regulatory sequences, the DNA sequences which control when a gene is turned on or off. "The rationale for comparing the genomes of different animals to identify
those sequences that are important is based on the understanding that
today's different animals arose from common ancestors tens of millions
of years ago," Rubin explains. "If segments of the genomes of two different
organisms have been conserved (meaning the sequences are the same in both)
over the millions of years since those organisms diverged, then the DNA
sequences within those segments probably encode important biological functions."
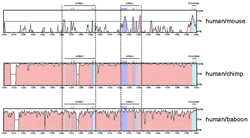
"Comparative genomics studies between evolutionarily distant species will readily identify regions of the human genome performing basic biological functions shared with most mammals," says Rubin. "However, it will invariably miss recent changes in DNA sequence that account for primate-specific biological traits." Rubin has likened comparisons between the human and mouse genomes to comparisons between an automobile and a go-cart: "Only the very basic parts and design features are similar." Whereas, he argues, comparing the human genome to that of a chimp or a baboon, is like comparing a sedan to a station wagon: "Nearly all the parts and design features are almost interchangeable." Until now, however, comparing the human genome to that of a chimp or baboon has been a problem since both genomes are so much alike. As Boffelli, who works with Rubin at both Berkeley Lab and JGI explains,
"There is only about a 5-percent difference between the human and the
baboon genomes. When you run comparisons between the two, all of the sequences
look just about the same. We can't distinguish function from non-functional
sequences." Rubin's research group at Berkeley Lab has been at the forefront of using
transgenic mice and the mouse genome to decipher the human genome and
to identify and study important genetic risk factors in the development
of human heart disease. He and his group believe that the ability to do
comparative genomic studies with non-human primates will prove especially
beneficial to human medical research. Their data from this study suggests
that sequencing the genomes of as few as four to six primate species in
addition to humans may be enough to identify much of the conserved functional
DNA sequences in the human genome. Berkeley Lab is a U.S. Department of Energy national laboratory located
in Berkeley, California. It conducts unclassified scientific research
and is managed by the University of California. Visit our Website at www.lbl.gov/. Additional information Dr. Edward Rubin can be reached at (510)486-5072 or Additional information can be obtained at The paper can be read at: |
|||||||||||||||||