| March 31, 2005 | science@berkeley lab | | lab a-z index | lab home |
|
|||
| A New Guide to Exploring the Protein Universe | ||||||||||||||||||||||||||
| Contact: Lynn Yarris, lcyarris@lbl.gov | ||||||||||||||||||||||||||
| Parallel universes are the subjects of heated debate in cosmology, but there is a parallel universe located here on Earth, and the fabric of its existence is as critical to our own as the fabric of space and time.
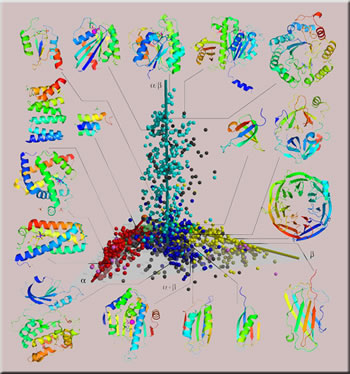
This parallel universe — which comes with its own Big Bang and expansion — is the universe of proteins, the vast assemblage of biological molecules that are the building blocks of living cells and control the chemical processes which keep those cells alive and functioning. It is estimated, based on the total number of known life forms on Earth, that there are some 50 billion different types of proteins in existence today, and it is possible that the protein universe could hold many trillions more. This points to a cornucopia of important new proteins waiting to be discovered. Given the enormity of the protein universe, however, how does a scientist know where to find the most densely populated (hence most promising) regions of protein space to explore? Help is on the way in the form of a comprehensive new 3-D map that brings order to the protein universe through a manageable organization. "We have constructed a protein-structure space map (SSM) based on
the distribution in 3-D space of the 1,898 known unique protein structures,"
says Sung-Hou Kim, a chemist who holds a joint appointment with Berkeley
Lab's Physical Biosciences Division and UC Berkeley's Chemistry Department.
"Because proteins with similar structures and functions are clustered
together in the SSM, when the structure of a new protein is first identified
it can be placed in the appropriate location on the map to reveal its
neighbors and its evolutionary history. This information can then be used
to predict the protein's function."
As the DNA base pairs that make up the genomes of a growing number of different organisms are sequenced, the next horizon for the biosciences is to identify coding genes and the molecular and cellular functions of the proteins encoded by them. Coding genes are DNA sequences that translate into sequences of amino acids which fold into proteins. The prevailing scientific method for predicting the function of a newly discovered protein has been to compare the sequence of its amino acids to the amino acid sequences of proteins whose functions have already been identified. A major problem with relying exclusively on this approach is that while two proteins may have similar structure and function, the sequences of their amino acids may be dramatically different. "From the data available at this time, it would seem that protein structure has been much more conserved during evolution than genetically based amino acid sequences," Kim says. "Because the functionally important portions of proteins fold into specific structures to perform their functions, structure-based functional inference can be used to characterize close relationships between different proteins that would be impossible to detect by using amino acid sequences alone, or by the similarity of whole structures." Through the eons, proteins have selectively evolved into the architectural structures best suited to do their specific jobs. These structures essentially stay the same for proteins from all three kingdoms of life — bacteria, archaea, and eukarya — even though the DNA sequences coding for a specific type of protein can wildly vary from the genome of one organism to another, and sometimes even within the same organism. Evidence of this conservation can be seen in the fact that, while the protein universe may encompass trillions of different kinds of proteins, most structural biologists believe there are probably no more than ten thousand distinctly different types of architectural structure motifs. "Each year the number of newly identified protein structures has increased exponentially, while the number of new protein folds has grown smaller," Kim says. "This and other observations strongly suggest that the total number of protein folds is dramatically smaller than the number of protein types." The SSM that Kim and his colleagues Jingtong Hou, Se-Ran Jun, and Chao Zhang have developed is based on protein structural data obtained from the PDB.Select database, a subset of the Protein Data Bank, the worldwide repository for data on 3-D biological macromolecular structures. Unlike the parent database, PDB.Select contains no redundant protein structures.
To construct the SSM, Kim and his colleagues used a mathematical technique called multidimensional scaling. Each of the 1,898 individual protein structures is represented by a sphere, which is assigned a color according to the widely used Structural Classification System of Proteins (SCOP); the spheres are distributed along three elongated regions, which are centered around three axes, denoted alpha, beta, and alpha-slash-beta. The results were published in the March 8, 2005 edition of the Proceedings of the National Academy of Sciences. In comparing the ability of the SSM to predict protein function to that of the most widely used current predictive method, the DALI Z scores, Kim and his colleagues found that the SSM method can often be used to predict the molecular function of new proteins whose function could not be predicted using the DALI Z scores. "The SSM was able to reveal similar functions even between proteins that had dissimilar structures and sequences," Kim says. "This observation suggests a scheme to infer protein functions based on the distances in the SSM, especially for proteins with new folds." The SSM should prove enormously beneficial as a guide to future explorations of the protein universe. It should make it possible for researchers to quickly classify newly discovered proteins and provide a running start in identifying the functions of these proteins. The SSM should also be a big help in developing rational strategies for the discovery or design of new, safer, and more effective pharmaceutical drugs. Most of these drugs work by interfering with or enhancing the function of a targeted protein. With the SSM, the search for drugs that act on a specific protein with fewer side effects will be made easier. Says Kim, "The SSM can be used to identify which other proteins with similar structures might also be affected by a given drug." Development of the protein universe SSM was funded by grants through the National Science Foundation and the National Institutes of Health. Additional information
|
||||||||||||||||||||||||||
| Top | ||||||||||||||||||||||||||