|
||
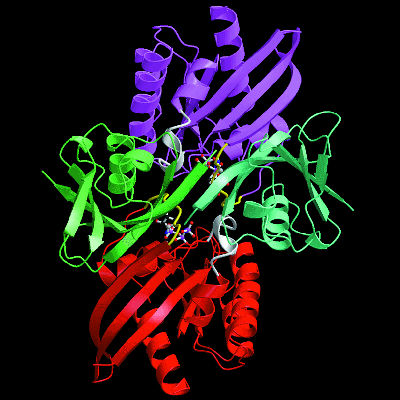
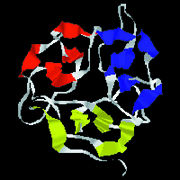
| Function follows form in proteins, so to learn what a protein does scientists must determine its physical shape. Many proteins are formed from distinct subunits called "domains" (shown here in violet, green, red, and teal), each of which contributes a specialized function to the overall protein. | ||
 |
|
 |
|
 |
|
 |
|
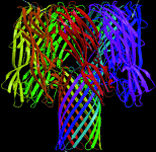
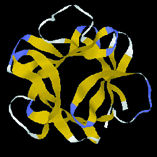
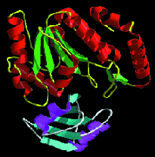
| Protein shapes reveal recurring structural motifs called "folds" that help define physical and chemical properties. Commonly seen folds include beta-sheets or ribbons (top two images), alpha-helices (third from top), and complex globular conformations (bottom). | |
Computational models of protein folding are providing valuable information
on three-dimensional structures, but for the precise requirements of protein
engineering and rational drug design, there is no substitute for high-resolution
3-D imaging. There are several approaches, each with its own special advantage,
but the workhorse technique for imaging protein structures is x-ray crystallography.
A Scattering of X-rays
When a beam of x-rays is sent through a crystal, the atoms in the crystal
cause the x-rays to scatter, creating a diffraction pattern. This diffraction
pattern can be translated by computer into 3-D images of the crystal.
Throughout its first 50 years, x-ray crystallography of proteins proceeded
at a tortoise pace. Collecting complete data sets for a single protein
crystal could take months or even years largely because laboratory x-ray
tubes don't produce enough photons in their beams. All that changed with
the arrival of synchrotrons designed expressly for the extraction of light
from accelerated electrons. When a beam of electrons accelerated to relativistic
(near light) speeds is forced to travel along a curved path, it emits
photons-copious quantities of photons. The energy and wavelengths of these
emitted photons are a product of the energy of the electron beam and the
curve of its path.
Berkeley Lab's Advanced Light Source (ALS) accelerates electrons to approximately 1.5 billion electron volts of energy, then stores them inside a ring that is 200 meters in circumference. An armada of focusing magnets holds the electrons in a hair-thin beam, and a series of bending magnets steers them around the ring, causing them to throw off strobe-like flashes of x-ray light as they move through the curves. The storage ring is also equipped with special magnetic "insertion" devices called "wigglers" and "undulators." Essentially a line of powerful magnets arranged for alternating polarity, an insertion device oscillates the path of the electron beam at the precise amplitudes and frequencies needed to generate beams of x-ray and ultraviolet light that are exceptionally high in flux (the number of photons) and collimation (parallel alignment of the photons).
 |
||
| Researcher Mhairi Donohoe at Beamline 5.0.2 of the MCF/ALS, a premier x-ray crystallography research facility that provides "hard" x-rays (3.5 to 14keV) and is optimized for imaging proteins and other biological molecules. | ||
Macromolecular Crystallography Facility
The ALS' Macromolecular Crystallography Facility (MCF) currently consists of a single beamline--5.0.2-running off a 38-pole wiggler insertion device that produces x-rays ranging in wavelengths from 0.9 to 4.0 angstroms and in energies from 3.5 to 14 keV (thousand electron volts). The higher end of this energy range-called "hard" x-rays-was supposed to be beyond the reach of the ALS, but engineering in the storage ring exceeded expectations. Increased photon energy means increased penetration, another important asset for protein crystallography. Precise tuning of the light also makes it possible for researchers at the MCF to use MAD-"multiple-wavelength anomalous diffraction"-an x-ray crystallography technique that is ideal for imaging proteins as well as other biological molecules.
Experimental activities at the MCF are led by Thomas Earnest, a biophysicist with Berkeley Lab's Physical Biosciences Division. Himself an expert in protein crystallography, Earnest heads a research group that is investigating proteins involved in signal transduction across cell membranes. The challenge of working with membrane proteins, which are especially difficult to crystallize-as well as being weak diffractors-keeps Earnest attuned to the needs of other protein crystallographers.
 |
||
| Biophysicist Thomas Earnest, who now oversees activity on the Macromolecular Crystallography Facility at the Advanced Light Source, was in charge of the facility's development. | ||
"One of our biggest strengths is that we run a total scientific program here," Earnest says. "We offer our users faster, higher quality data over a wider dynamic range, and we offer them a choice of crystallographic techniques with fast data collection."
The benefits to be reaped from MCF's state-of-the-art protein crystallography capabilities are evident in two recent experiments that received considerable attention from Science and Nature, the science world's premier journals. In one, a team of researchers from the University of California's Santa Cruz (UCSC) campus, working with Earnest, produced the first high-resolution images of a complete ribosome, the cell organelle that has been called a "protein factory" because it is responsible for protein synthesis. In the other experiment, a team of Berkeley Lab and UC Berkeley researchers, again working with Earnest, produced the first three-dimensional look at a member of a large family of proteins that plays a central role in the development of cystic fibrosis and can also block the therapeutic effects of medications.
A Ribosome Complete
 |
||
| Science magazine named the 7.8 angstrom resolution image of the 70S ribosome, the first of a complete ribosome, as one of the top ten scientific breakthroughs of 1999. The image, recorded at the ALS, revealed that 70S consists of two asymmetric domains, 30S (pink) and 50S (aqua). | ||
UCSC molecular biologist Harry Noller led the ribosome experiment which, among other accomplishments, demonstrated that there is much more to ribosomal structures than had been gleaned through earlier indirect or low-resolution observations. MCF images of the 70S ribosome of the bacterium Thermus thermophilus at a resolution of 7.8 angstroms revealed an RNA-protein bridge spanning the two asymmetric "domains" that make up bacterial ribosomes-a domain being a distinct substructure of a protein. Preliminary work indicated that this RNA-protein bridge is the basis for communication between the two domains of Thermus thermophilus.
"One gets the impression (from the bridge image) that there are systems of long-range communication connecting distant parts of the ribosome," Noller says.
Other research groups have obtained high-resolution images of individual ribosome subunits, but this marked the first time a detailed image of an entire ribosome complex was obtained. Ribosomes receive and somehow unite "messenger RNA" molecules from the nucleus with "transfer RNA" molecules from the cytoplasm. Messenger RNA carries the genetic code for assembling proteins; transfer RNA carries the amino acids from which proteins are made. A detailed understanding of ribosomal structures would be a giant step toward understanding the mechanism by which these critical organelles function. However, until the advent of synchrotron radiation sources such as the ALS, obtaining this information was a challenge deemed insurmountable. Although smaller than most viruses, a ribosome is a very large macromolecular complex, consisting of three RNA and more than 50 protein molecules.
"Obtaining atomic-resolution diffraction data for so large a macromolecular complex can only be done with a high-brightness source of x-rays," says Earnest. "Our facility is one of the best in the world for this work."
The 70S ribosome images obtained at the MCF gave Noller and his colleagues some ideas as to how transfer RNA interacts with the ribosome, and how the two ribosomal subunits interact with each other. In both cases, there appear to be complex networks of molecular interactions criss-crossing the ribosome, often involving interactions with a third type of RNA-called ribosomal RNA.
"Our images suggest very strongly that the ribosome is a very complex
machine with many moving parts," says Noller. "Our images also make it
also clear that most of the excitement of figuring out the molecular mechanisms
behind this machinery lies ahead."
ABC's of Cystic Fibrosis and Protein Folding
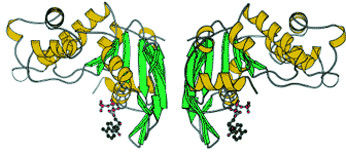
Sung-Hou Kim, a chemist who holds a joint appointment with Berkeley Lab's Physical Biosciences Division and UC Berkeley's Chemistry Department, was co-leader of an experiment at the MCF in which the 3-D structure of a protein called HisP was solved. HisP belongs to a family of proteins that function as "engines" for a larger group of protein complexes known as ATP-binding cassette (ABC) transporters which are responsible for carrying substances back and forth across the inner membranes of cells. Among the many medically significant proteins in the ABC transporter family are the cystic fibrosis transmembrane regulator (CFTR) and a multidrug resistance protein (MDR) called P-glycoprotein.
 |
|
| ABC transporter proteins such as this are responsible for carrying substances across the inner membranes of cells. ABC proteins play a major role in the development of cystic fibrosis and can also impede the medicinal effects of therapeutic drugs. | |
Cystic fibrosis is the most common fatal genetic disease in the United States today, occurring in approximately one of every 3,300 live births. It is caused by mutations in the CTFR gene that result in defective CFTR proteins. MDR proteins are the bane of the medical community because they counteract the effects of pharmaceutical drugs, forcing doctors to increase prescribed dosages in order to obtain desired results.
"Cystic fibrosis occurs when the ABC transporters are not working properly, and multidrug resistance occurs when ABC transporters are working too well," says Kim. "With our 3-D crystal structure, we have provided a structural basis for understanding the engine functions of ABC transporters, and this knowledge could be used to better understand and perhaps treat cystic fibrosis or to design ways to inhibit multidrug resistance."
The spectral quality of ALS x-rays in combination with the instrumentation at the MCF enabled Kim and his colleagues to resolve details down to 1.5 angstrom resolution in their images of a HisP protein from an E.coli-like bacterium known as Salmonella typhimurium. ABC transporters contain two domains which bind to ATP (adenosine triphosphate), the molecule that serves as a sort of traveling battery pack for the cell, delivering energy wherever it is needed. ATP-binding domains are thought to power the molecular machinery of two other domains that span a cell's membranes (hence form the connection between the interior and exterior of the cell). Scientists would really like to know what these membrane-spanning domains look like and how the ATP-binding domains power them. Solving the HisP protein structure is a critical step towards this goal.
For all the potential importance that solving this HisP protein structure holds for cystic fibrosis research and rational drug design, the fact that Kim and his group were subsequently able to correlate its structural details with the biochemical, genetic, and biophysical properties of the wild-type and several known mutant HisP proteins could have even more significant consequences for an emerging area of science now referred to as "structural genomics."

|
||
| Sung-Hou Kim (left), working at the Advanced Light Source with Li-Wei Hung, Tom Zarembinski and Jochen Mueller-Dieckmann, solved the 3-D structure of a hypothetical protein and then correctly predicted its function. | ||
By combining the determination of protein structures with the identification of the protein-coding DNA sequences in the genome of a given organism, structural genomics seeks to learn the functions, through images and models, of all the proteins encoded in completed genomes. Since understanding the molecular (physical and chemical) functions of proteins is required to understand their cellular functions, the advancement structural genomics promises enormous ramifications for all the fields of biology, especially biomedical research. The scope of the structural genomic challenge is so monumental, however, it is far beyond daunting.
"There are simply too many genes to determine the protein structures for all of them," says Kim.
Meeting
The Structural Genomic Challenge
In response to this challenge, or, more accurately, as a way around it, Kim and other crystallography leaders have proposed that rather than even try to determine the 3-D structure of every single protein, scientists should instead target the recurring structural motifs or folds underlying all protein architecture. Once these fundamental structural motifs-called a "fold basis set"-have been identified and categorized on a database, they could serve as a basis for predicting the functions of newly discovered proteins. Though still a challenge, this would be a more manageable undertaking because while the different protein types may number in the hundreds of thousands, most biologists agree there are probably fewer than ten thousand distinctly different types of folds. Explains Kim, "A smaller number of new protein folds are discovered each year despite the fact that the number of structures determined annually is increasing exponentially. This and other observations suggest strongly that the total number of protein folds is substantially smaller than the number of genes, and a majority if not all proteins may belong to a fold basis set."
 |
|
 |
|
 |
|
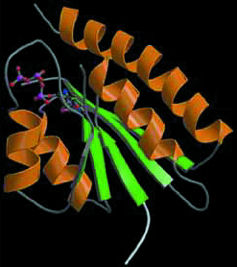
| X-ray crystallography at the ALS has been successfully applied to structural genomic research involving a microbe called Methanococcus jannaschii (MJ). Top image is an electron-density map of MJ protein 0577 showing a bound ATP (yellow stick structure). Middle image shows a tertiary structure of MJ0577 that is a nucleotide binding fold. Bottom image is a 3-D model of a small heat-shock protein made from a cloned MJ gene. | |
To identify these fold basis sets, Kim argues for working with a representative sample of protein populations to be obtained from organisms whose entire genomes have been sequenced, the rationale being that through the eons, families of proteins have selectively evolved into the structural shapes best-suited to do their specific jobs. These shapes essentially stay the same for proteins of a given function in all three domains of life-bacteria, archaea, and eukarya-but the DNA sequences encoding for proteins with the same function can greatly vary from the genome of one organism to another. This is why just knowing a protein's sequence doesn't always tell you everything that protein might do, but knowing a protein's folding structure will most likely point you in the right direction.
Recently, Kim and his research group solved the structure of a "hypothetical protein" (a protein coded by a gene with no known function based on its DNA sequence) from Methanococcus jannaschii, an archaean microbe that lives in deep-sea vents where the temperatures climb to about 100 degrees Celsius, and found it contained ATP. They compared their structure to the structures of known proteins in the Protein Data Bank, the international repository for all the known protein structures. On this basis, they deduced that the hypothetical protein functions as a molecular switch for activating or de-activating other proteins.
"Our structural data gave us a lead as to the molecular function of the hypothetical protein function which we were able to verify through biochemical tests," says Kim. Last year, with funding from the U.S. Department of Energy, Kim and his research group began a pilot structural genomics study. Again, they worked with proteins from Methanococcus jannaschii, whose entire genome has been sequenced and found to hold 1,738 genes. These genes are readily introduced into bacteria for mass production of their proteins, and the proteins themselves, coming from a microbe that thrives in hot environs, are heat-resistant to the rigors of purification and crystallization. Furthermore, Methanococcus jannaschii is a "deeply-rooted organism," meaning it is one of the most primitive of all life forms.
"Since Methanococcus jannaschii was on the ground floor of evolution, the information we obtain on its protein folds should be transferable to the proteins of other organisms," says Kim.
To date, Kim and his research group, working at the MCF, have cloned about 50 different proteins from Methanococcus jannaschii and have determined the 3-D structures of eight of them, four of which are hypothetical. Some of these protein structures display folding patterns never before reported, showing that he and his colleagues are on the right track. "There is a clear and compelling role for protein crystallography in providing a foundation for structural genomics," Kim says. "Given that the use of synchrotron radiation can dramatically decrease the time required to solve a novel structure, the need for synchrotron radiation facilities is not to be underestimated."
More
Beamlines, Faster Throughput
The Protein Structure Initiative, under the auspices of the National Institute of General Medical Sciences, was prompted in part by the success of the work with Methanococcus jannaschii by Kim and his group, along with comparable work by other groups with other organisms.
This initiative, coupled with the flood of new genomic data from the Human Genome Project pouring into the public databases, is expected to dramatically boost the demand for beamtime at synchrotron-based crystallography facilities. As part of the ALS, which has been designated a "national user facility" by the U.S. Department of Energy, the MCF's resources are open to all qualified users. However, the demand for beamtime by would-be users is already so high the MCF has only been able to grant about 35 percent of all the requests received. To help meet current demands and the surge that will be coming, the MCF is adding two more experimental beamlines-5.0.1 and 5.0.3-which are scheduled to start operating in the summer of 2000. These two new beamlines will provide monochromatic beams of x-rays at 12.4 keV of energy. Even though they won't offer the MAD capabilities of the existing beamline, the availability of these new beamlines, each with three experimental hutches for simultaneous research, will relieve some of the user pressure on the MCF.
"There are plenty of experiments that don't require MAD," says Earnest, "but right now, beamline 5.0.2 is being used for everything, monochromatic as well as MAD."
More help for protein crystallographers will come toward the end of the year 2001 when the ALS is scheduled to replace three of the bending magnets now in its storage ring with powerful new superconducting magnets. Called "superbends," these new magnets will tighten the path of curvature of the electron beam as it circles through the ring, yielding hard x-rays perhaps as high as 50 keV in energy. There could easily be three new crystallography beamlines attached to each of these superbends bringing the total number of ALS protein crystallography beamlines to a dozen by the year 2002. This would make the ALS one of the largest synchrotron sources for structural biology research in the world.
More beamlines alone will not be enough to meet the growing needs of crystallographers. Faster throughput -the time required to produce and set up protein crystals in the beamline, illuminate them with x-rays, and collect the data-is also required. The answer to faster throughput, Earnest says, is automation.
"Wherever human hands touch the crystals is where the bottlenecks arise," he says. "We really want to minimize the amount of human intervention that is necessary."
Earnest would like to automate the entire crystallography process, from start to finish, which means from the front-end work of growing the crystals (see story on page 18) to the down-stream work of collecting and analyzing the data, to the final stage of entering the results in a public database.
"With current techniques, it will take decades to amass a meaningful collection of data because we just can't solve crystal structures fast enough," Earnest says. "But with full automation, we should be able collect complete data sets on 10 to 15 crystals per day on each of our beamlines."
More synchrotron radiation beamlines plus higher throughput add up to a bright future for protein crystallographers, and this in turn will brighten prospects for all of biology. As Bruce Alberts, renowned biochemist and President of the National Academy of Science once wrote:
"The great future in biology lies in gaining a detailed understanding of the inner workings of the cell's many marvelous protein machines." - end -
| < Research Review | Top ^ | Next > |