BERKELEY — In a few seconds an origami artist can fold a sheet of
paper into a bird or flower or pagoda or other intricate shape. In much less time a string
of amino acids can fold itself into a protein, the kind of molecule that comes in many
thousands of complex shapes and does most of the work of life. Origami can be taught, but
no one knows how proteins fold themselves so quickly into the same shapes virtually every
time.
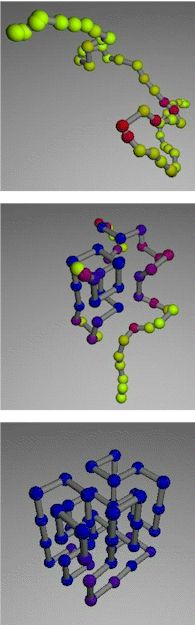 Top to
bottom: unfolded, intermediate, and stable states of a model polymer
|
Now, computer models devised by Daniel Rokhsar and his colleague Vijay Pande of the
Department of Energy's Lawrence Berkeley National Laboratory, working at the National
Energy Research Scientific Computing Center (NERSC), have revealed unexpected regularities
in the pathways of protein-like structures. They report their findings in the Proceedings
of the National Academy of Sciences, February 16, 1999 (vol. 96, no. 4).
"We're interested in the physical mechanisms by which biomolecules achieve their
structures," says Rokhsar, who is head of the Computational and Theoretical Biology
Department in Berkeley Lab's Physical Biosciences Division and a professor of physics at
the University of California at Berkeley.
The precise structure of biomolecules often reveals their functional secrets, a fact
that became clear in the early 1950s when Linus Pauling solved the alpha-helix structure
of keratin protein and Watson and Crick solved the double-helix structure of DNA.
Protein shapes determine everything from the texture of hair and horn to the catalytic
coupling and uncoupling of innumerable enzymes essential to keep life's processes humming.
Misfolded proteins can cause disease; in humans, sickle-cell disease and other anemias,
for example, are caused by the misfolding of hemoglobin, whose normal structure,
resembling a miniature spring-clip, allows it to capture, transport, and release oxygen in
the bloodstream.
For any protein there is a "native state conformation," a thermodynamically
most-stable structure that depends upon the energy of the bonds that form when the acid
residues come close together, and upon whether a given group of amino acid residues is
hydrophobic or hydrophilic, and so on. But newly manufactured proteins are far from their
native state.
Proteins are punched out rather like ticker-tape by ribosomes that add amino acids one
at a time. Although the order of the amino acids is ultimately specified by a length of
DNA (the gene for that protein), how the order specifies the protein's distinctively
folded structure and directs pathways to that structure is not yet understood.
"Protein chains all fold differently—even proteins of the same kind fold into
their final state by sampling many different conformations—because they start from
different initial states," says Rokhsar. "Yet somehow they start from an
unfolded state and achieve the folded structure quickly, reliably, and reversibly."
To demonstrate the magnitude of the challenge, Rokhsar suggests contemplating a single
node—a stand-in for a single amino acid residue—represented by a ball on the end
of a stick. "Let's limit to five the directions the next stick-and-ball can
extend—right, left, up, down, or straight ahead," says Rokhsar. "If there
are five links in the chain, that's five to the fifth ways the chain could fold, 3,125
possibilities. If there are a hundred links in the chain—not unusual for a
protein—there would be something like 10 to the 30th possibilities. If you tried them
randomly, even a trillion times a second, it would take longer than the age of the
universe to get the right structure."
Rokhsar and Pande, who is a Miller Postdoctoral Fellow at UC Berkeley's Department of
Physics, approached the problem by designing a protein-like model heteropolymer of 48
units whose properties define a stable "native structure"—a compact lattice
in three dimensions with each bend at a right angle, resembling a jungle gym made of
Tinker Toys.
Using the Cray T3E computer at NERSC, Rokhsar and Pande repeatedly unfolded the model
by raising its (simulated) temperature, then lowered the temperature and watched it refold
itself. For each folding sequence they separately tracked the position of each of the 48
"mers," the units equivalent to a protein's amino acid residues.
Even with a model far less complex than most real proteins, the number of possible
initial conformations is astronomically large, and each path to stability is virtually
unique. By sampling the state of the writhing polymer every 6,000 iterations—taking a
single-frame snapshot of the shape—the researchers made movies that showed the model
polymer seeking and eventually finding its stable state. Typically some three-quarters of
a million iterations were required before the model polymer stabilized.
The average position change of each unit was recorded from frame to frame, and the rate
of change was color-coded—from yellow for units that thrashed continually, through
the spectrum to blue for those that held still, at least temporarily. This data could be
arranged in "fluctuation smears" to give a cumulative picture of the position of
the units at any moment in the process.
Remarkably, Rokhsar and Pande discovered common features among the numerous folding
pathways. At first the unfolded polymers fluctuated wildly through several hundred
thousand configurations—then suddenly settled into a partially folded intermediate
state, in which a stable core structure was accompanied by flailing loops and dangling
ends. After another couple of hundred thousand iterations, the polymer abruptly locked
into its native state.
These sudden transitions are evocative of phase changes, like the changes from a gas to
a liquid to a solid. There are distinct classes of intermediate states for the model
polymer, however, which correspond to different groups of units that temporarily achieve
stability during the intermediate phase. Each class of intermediate states represents a
set of related pathways from the unfolded to the native state.
When Rokhsar and Pande repeated their simulations with model polymers of 64 and 80
mers, the folding pathways also grouped themselves into separate classes of intermediate
states.
These intermediate phases are closely analogous to partially unfolded states (PUFs)
which have been observed in real proteins, as well as to intermediate states inferred to
exist in other real proteins. It is likely that knowledge of PUFs, plus inferences about
similar
phases from other protein studies, can predict transition states of some kinds of
proteins in the real world. Rokhsar's and Pande's discovery of well-defined transition
states in model-polymer folding has important implications for the development of a
general theory of protein folding. Verifying these results using models with atomistic
detail is the next important step.
The lattice model of protein folding can be seen in spectacular action by visiting http://hubbell.berkeley.edu/nsb.html.
Also see the Pande Group
website and the gallery on that
site.
