| lab a-z index | phone book |
| May 29, 2008 | news releases | receive our news releases by email | science@berkeley lab |
|
|||
|
|||
The Structure of XPD Sheds Light on Cancer and AgingThe role of a unique DNA-repair protein in multiple diseases |
||||||||||||||||
| Contact: Paul Preuss, (510) 486-6249, paul_preuss@lbl.gov | ||||||||||||||||
BERKELEY, CA — The protein XPD is one component of an essential repair mechanism that maintains the integrity of DNA. XPD is unique, however, in that pinpoint mutations of this single protein are responsible for three different human diseases: in xeroderma pigmentosum, extreme sensitivity to sunlight promotes cancer; Cockayne syndrome involves stunted growth and premature aging; trichothiodystrophy, characterized by brittle hair and scaly skin, is another form of greatly accelerated aging. Now, for the first time, researchers at the U.S. Department of Energy's Lawrence Berkeley National Laboratory and the Scripps Research Institute have solved the essential structure of XPD. The structure reveals how discrete flaws in the remarkable architecture of XPD — as seemingly insignificant as a change in either of two adjacent amino acid residues — can lead to diseases with completely different phenotypes, and gives novel insight into the processes of aging and cancer.
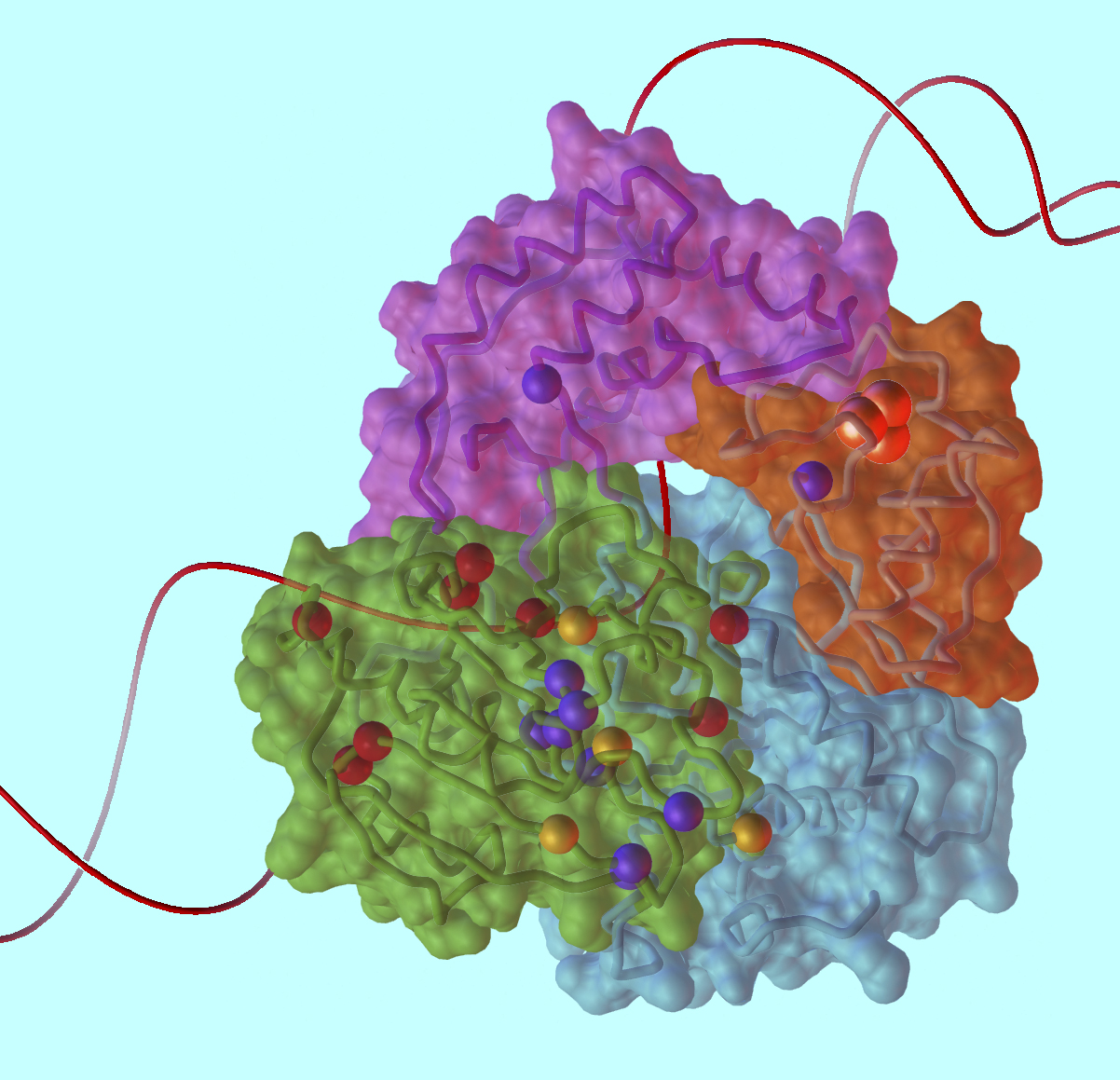
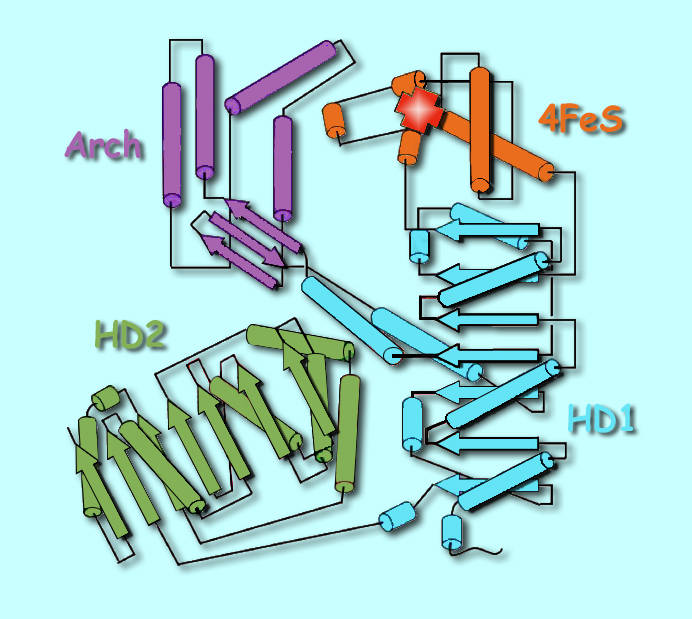
The team that solved XPD's structure was led by John Tainer, a professor in the Department of Molecular Biology and the Skaggs Institute of Chemical Biology at Scripps and a visiting scientist in the Life Sciences Division (LSD) at Berkeley Lab. "With this exciting work we have helped open the door to understanding how molecular interactions and activities in the cell result in aging — which is too much cell death — or in cancer, where defective cells are kept alive," Tainer says. Li Fan of the Scripps Molecular Biology Department performed the x-ray crystallography of XPD at DOE's Stanford Linear Accelerator Center and the Advanced Light Source at Berkeley Lab. The biochemistry of XPD was assessed by Jill Fuss, a biochemist in Priscilla Cooper's laboratory in the Genome Stability Department of Berkeley Lab's LSD, and by Quen Cheng, a Research Associate in the Cooper lab. The team also included Andrew Arvai of Scripps, Michal Hammel of Berkeley Lab's Physical Biosciences Division, and Victoria Roberts of the San Diego Supercomputer Center at the University of California at San Diego. The researchers report their results in the May 30 issue of the journal Cell. Crystallizing XPD"XPD is one of a group of genes originally identified because mutations in it were found in the cells of some xeroderma pigmentosum patients — which is where the initials XP come from — and of certain Cockayne syndrome patients as well," says LSD's Cooper, who has long studied the molecular basis of DNA repair and how its failures contribute to these diseases. "The exact roles of the XP proteins in carrying out DNA repair were only discovered much later." XPD is a DNA unwinding machine in the TFIIH molecular factory, which along with other XP proteins performs the kind of DNA repair called nucleotide excision repair. The TFIIH complex also functions in normal gene transcription and transcription-coupled repair. Nucleotide excision repair involves removing and replacing a strand of DNA that contains one or more damaged bases, damage that can be recognized because it distorts the helical structure of double-stranded DNA. Once the damage is located, TFIIH is brought to the site and unwinds the helix to expose a 30-nucleotide "bubble" in the DNA around the damaged region. XPD, a helicase, is one of two proteins in TFIIH that does the helix unwinding. Next, TFIIH's partner proteins XPG and XPF, nucleases that cleave nucleotides apart, clip out the damaged section of the strand. A polymerase, which joins nucleotides to one another, rebuilds the correct sequence by using the opposite strand as a template. Finally the nick is sealed by a ligase, which joins the ends of DNA strands together. Although not found in all organisms (bacteria use different proteins to carry out a similar DNA repair process), the gene for XPD, like many others essential to the basic processes of life, is highly conserved among numerous species, from humans to one-celled Archaea. The gene sequence has long been known, but the structure of the protein has eluded researchers, partly because XPD is so hard to purify and crystallize. "We first tried the human protein, but it's very difficult to express as a soluble recombinant protein," says Jill Fuss. "It turns out that human XPD is quite insoluble." Getting enough to crystallize means making quantities of it in a soluble form, she explains, using recombinant E. coli bacteria or other systems. "So we went searching for homologs," corresponding proteins from other organisms; Fuss notes that in a situation like this, "what crystallographers commonly turn to are thermophilic organisms" — heat lovers, like microorganisms that thrive in boiling hot springs, or worms that flourish near undersea volcanic vents. "Because they live at high temperature, their proteins are generally more stable." The Archaea Sulfolobus acidocaldarius lives happily at 80 degrees Celsius and pH 3 — equivalent to a vat of hot acid — and its XPD protein is indeed stable. Although it lacks the human protein's extensions, the core of S. acidocaldarius XPD is similar to that of human XPD — 22 of the 26 locations in the human gene sequence where mutations associated with disease occur are also found in the archaeal protein's catalytic core. A unique feature of XPD and a few other helicases is a group of iron and sulfur atoms that can eagerly bind with oxygen; to prevent oxidation (what might be called "rust"), the purified protein had to be crystallized inside an anaerobic glove box. "After growing hundreds of crystals, we knew we were on the right track when the crystals were a brownish-red color, which meant that the iron-sulfur cluster was intact," says Li Fan, formerly a fellow at Berkeley Lab and currently a researcher at Scripps. The structure of XPDTwo versions of the protein were needed to do x-ray crystallography: the native conformation and a variant in which all of its methionine amino acid residues were replaced by selenomethionines. In the synchrotron's beam the heavy selenium atoms signal their presence and allow unambiguous registration of the protein's methionines in proper sequence. The structure of XPD came as a surprise. Of the protein's four domains (distinct substructures), two are helicase motifs, HD1 and HD2; another is the iron-sulfur domain, 4FeS, which protrudes in a way that implies an important role in opening DNA and sensing DNA damage. The fourth domain, called the Arch, was wholly unexpected. With 4FeS, the Arch domain forms a curious archway over a tunnel-like opening at the end of a long groove. Given the electric charge of the amino acids lining the groove, the topograpy suggests a passageway for channeling DNA. "If you stretched out the linear sequence of the protein, you would never guess that those different pieces fold together," says Cooper. Proteins result from folding linear strings of amino acid residues, but in XPD the folding is an intricate knot. One of the helicase domains, HD1, takes portions of its sequence from separate regions of the linear string, and the Arch and 4FeS domains sprout from different parts of HD1.
The complex topography of the protein, and the specific amino acid residues of which it is formed, suggest the specialized functions of different structural features. The helicase domains are designed for flexible movement; the channels seem well suited to binding DNA, or the ATP molecules that when hydrolyzed provide the energy for unwinding; and the oxidation-sensitive 4FeS domain is strategically placed for detecting DNA damage. Structure, function, and diseaseTo assess XPD's biochemical activity in terms of its structure and to learn how even single-residue changes in structure can derail the process, "We moved back to the test tube," says Fuss. "There are three activities we can measure: one is ATPase activity, its ability to hydrolyze ATP; another is DNA helicase activity, its ability to unwind duplex DNA; and finally its DNA binding activity." Says Cooper, "The thing that XPD does in the TFIIH complex, in the cell, is unwind DNA to allow nucleotide excision repair to occur — that's its critical function. Helicase activity measures that unwinding. And you can't have helicase activity if you can't bind to DNA in the first place, or if you can't hydrolyze ATP." These activities were measured using 15 mutant forms of XPD, designed with specific mutations known to be involved in xeroderma pigmentosum, Cockayne syndrome, or trichothiodystrophy. In all three diseases, patients are sensitive to sunlight — ultraviolet light is a chief cause of the nucleotide damage that TFIIH specializes in repairing. But while xeroderma pigmentosum patients are thousands of times more prone to skin cancer than the population as a whole, neither Cockayne syndrome nor trichothiodystrophy patients show an increase in skin cancer. Instead they exhibit different forms of accelerated aging. "Until you have structures of the components of TFIIH, it's difficult to understand precisely the effect of particular mutations," says Cooper. "In the case of XPD, there's been a mystery all along that mutations that are very close together — and in one case, even in adjacent amino acids — can cause different diseases. Says Fuss, "In that case the two amino acids are side by side in the linear structure, but because of the intricate folding of XPD they end up with very different functions, acting as DNA hooks or structural clamps." Cooper says, "It's only once you see the structure that you realize why mutating one or the other of those two adjacent residues produces different effects — one is right in the DNA binding channel, and the other is going to affect the movement of the domains relative to each other. A mutation in one causes xeroderma pigmentosum. A mutation in the other causes Cockayne syndrome." Cooper explains that this may be because the Cockayne syndrome-causing mutation hampers XPD's binding with its TFIIH partner proteins, causing TFIIH to act as if it's doing DNA repair when it should be doing transcription — and thus allowing the wrong incisions in the DNA. "Sometimes combining structures from crystallography with activities from biochemistry is like turning on a light in a dark room — we can suddenly see how XPD works," says Tainer. The mutations that cause xeroderma pigmentosum directly affect the channels in the protein that are predicted to bind DNA and ATP. These changes make the mutant XPDs less efficient at unwinding DNA. Other mutations, which result in Cockayne syndrome in combination with xeroderma pigmentosum, interfere with the flexibility of the helicase domains, which must be able to conform to the changing work site during repair and, in particular, bind with other proteins that are components of the repair machinery. Trichothiodystrophy is caused by mutations that may or may not reduce helicase activity but do interfere with "framework stability," the structural integrity of the protein. And some mutations in the 4FeS domain wreck XPD's structure to such a degree that helicase activity vanishes completely. Says Tainer, "The combined experiments showed that xeroderma pigmentosum mutations are binding defects; mutual xeroderma pigmentosum and Cockayne syndrome mutations are conformational defects that make the protein more rigid; and trichothiodystrophy mutations are framework defects that make the protein more floppy." Cooper adds, "An important part of understanding the effects of these mutations is knowing exactly how they bind to DNA, which has proved to be a particular challenge and one of the next goals of our research. Although we have computational predictions and tested them with biochemistry, we don't yet have a structure of XPD bound to DNA." The structure of XPD's catalytic core represents a major advance in the understanding of the TFIIH repair and transcription machinery and yields immediate insights into how different breakdowns in this machinery give rise to the physical signs of aging, and to cancer. There is much more still to learn about this remarkable protein. Structural and biochemical research on XPD was made possible at Berkeley Lab through a combination of programs. The SIBYLS beamline at the ALS is supported by DOE. Sulfolobus proteins were characterized in DOE's Genomics: GTL Program. Funding from the National Cancer Institute has supported understanding of DNA repair machines. "XPD helicase structures and activities: Insights into the cancer and aging phenotypes from XPD mutations," by Li Fan, Jill O. Fuss, Quen J. Cheng, Andrew S. Arvai, Michal Hammel, Victoria A. Roberts, Priscilla K. Cooper, and John A. Tainer, appears in the 30 May, 2008 issue of Cell and is available here, or online at http://www.cell.com. Berkeley Lab is a U.S. Department of Energy national laboratory located in Berkeley, California. It conducts unclassified scientific research and is managed by the University of California. Visit our website at http://www.lbl.gov. |
||||||||||||||||
| Top | ||||||||||||||||