-
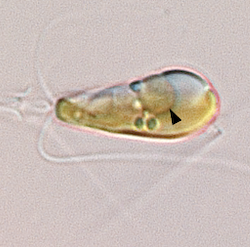
Scientists Discover Nitrogen-fixing Organelle

Two Scientists Join Ranks of AAAS Fellows

Toward Engineering Cell-like Factories

Cracking Sugarcane’s Genetic Code

Commemorating Judy Campisi
About Biosciences
Enabled by Berkeley Lab’s world-class user facilities and complementary research programs, our scientists and engineers contribute groundbreaking discoveries and innovative solutions to complex scientific and societal challenges.
Our Science
We advance biological science relevant to national-scale challenges in energy, environment, health, biomanufacturing, and technology development.

Our People
We collaborate across disciplines to develop innovative solutions that arise from a diversity of thought, approaches, experiences, and roles.

Divisions & User Facility
Biological Systems & Engineering (BSE)
BSE investigators lead efforts that combine the power of biology with the tools of engineering to advance sustainable energy, develop biomanufacturing solutions, and improve human health.

Environmental Genomics & Systems Biology (EGSB)
Scientists in EGSB aim to link genome biology to ecosystem dynamics. They develop systems-level models using genomics, integrated molecular observation, controlled manipulation of model organisms, and data science.

Molecular Biophysics & Integrated Bioimaging (MBIB)
MBIB researchers generate a predictive and mechanistic understanding of biological processes. Their goal is to harness basic cellular functions to solve national challenges in energy, environment, health, and biomanufacturing.

DOE Joint Genome Institute (JGI)
The JGI is a DOE National User Facility that provides genomic science services to researchers around the globe. Through their user support and discovery science, JGI researchers further DOE missions related to clean energy and environment.

Was this page useful?
Send

 Follow us:
Follow us:

