| November 5, 2004 | science beat | | lab a-z index | lab home |
|
|||
| Doing More with Less A new way to learn how large proteins move |
| Contact: Dan Krotz, dakrotz@lbl.gov |
| In a development that will help scientists better determine how many large proteins work, Berkeley Lab's Paul Adams and collaborators used the latest advances in computational analysis to study how a complex biological machine refolds proteins, a process critical to cell survival. They took advantage of the fact that the protein, called a chaperonin, encapsulates unfolded proteins by moving large portions of its structure in unison, like a hand clenching a marble. This means that only a few frames of crystallographic images, each revealing the protein at a different stage of its motion, are needed to picture the entire process. And because many large proteins are believed to behave this way, their work also means that the mountain of crystallographic data collected over the past ten years — much of which only portray large-scale changes in proteins — can help determine how many proteins twist and turn. "We can get more out of crystallographic experiments than we thought we could," says Adams, a staff scientist with Berkeley Lab's Physical Biosciences Division and the deputy principal investigator of the Berkeley Structural Genomics Center. "Although we often can't model the behavior of a protein atom by atom, it is possible to visualize how large portions of a protein's atoms move together." As of October, 2004, there were more than 27,000 structures in the Protein Data Bank, a worldwide repository of three-dimensional structures of large proteins and nucleic acids. Not all of these proteins lend themselves to this broad-brush analysis, because not all of them exhibit what's called rigid body movement, in which discrete groups of their atoms move as a single unit. But for those that do, Adams's technique could offer an efficient way to understand how they move. His team, which included Charu Chaudhry and Arthur Horwich of Yale University and Axel Brunger of Stanford University, developed the method using a chaperonin found in Escherichia coli called GroEL. Like similar molecules found in the cells of all organisms, GroEL repairs proteins that unfold due to adverse conditions such as heat stress. If too many proteins unfold, they cling together and cause cell damage, even death. To keep cells healthy, GroEL somehow surrounds a poorly folded protein, isolates it with a lid, then quickly stretches the protein before letting go, helping it move back to its correct shape. To visualize how GroEL conducts this rescue work, the team used crystallographic data representing the protein in four different configurations: with its lid off, with its lid on, bound to the energy-providing enzyme adenosine triphosphate, and immediately after it attempts to refold a protein. They then ran this data through newly developed software that calculates how groups of atoms within the protein rotate and translate around fixed points. This analysis revealed the direction in which each of the protein's parts, or rigid bodies, was moving at the time the data sets were taken. In this way, they were able to model GroEL's movement using only about 20 parameters per rigid body, as opposed to the thousands of parameters needed to model a protein's atom-by-atom movement. "We can see the protein's large conformational changes through these snapshots," says Adams. "The original crystal images are hugely informative, but we want to add to these static pictures information about how large groups of atoms move in the molecule." Their success marks one of the first times this method has been used to elucidate how parts of a large molecular machine move. As such, it offers a way to experimentally validate theoretically and computationally derived models of molecular motion. Adams believes the technique could also soon help reveal the dynamics of other molecular machines that possibly change shape via large, sweeping motions, such as the proteins that make up muscles or molecules that help DNA replicate. And to make the process more efficient, his group is developing an automated way to determine which atoms team up and behave like rigid bodies. "We could take a structure, figure out its rigid bodies, and then perform the analysis with the experimental data," says Adams. "Our overall goal is to take crystallographic data and extract information about large motions in big systems to better understand how they work." Additional information
|
| Top |
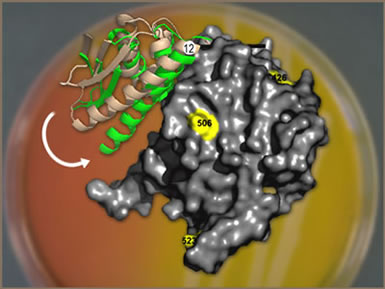 A few frames of crystallographic data can reveal how large portions of
a molecular machine, in this case the chaperonin GroEL, move. (Numbers
indicate labeled peptides, used as references to track the motion of the
protein's individual components.)
A few frames of crystallographic data can reveal how large portions of
a molecular machine, in this case the chaperonin GroEL, move. (Numbers
indicate labeled peptides, used as references to track the motion of the
protein's individual components.)