| February 17, 2004 | science beat | | lab a-z index | lab home |
|
|||
| Protein Crystallographers get Help From Elves | ||||||||||||||||||||
| Contact: Lynn Yarris, lcyarris@lbl.gov | ||||||||||||||||||||
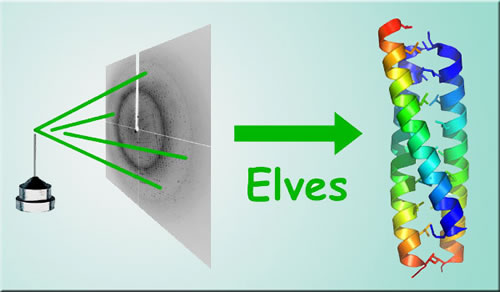
| As everyone who has seen the Lord of the Rings films or read J.R.R. Tolkein's works knows, when something had to be done quickly and efficiently, Middle-Earthers turned to the elves. Now, modern protein crystallographers may soon be doing the same. Elves is the name of an "expert system" of computer software programs that can automatically generate 3-D high-resolution images of proteins from x-ray diffraction data. The system was developed by a Berkeley Lab scientist working at one of the protein-crystallography beamlines of the Advanced Light Source (ALS).
"Elves decreases the time and training required to perform the computational steps of x-ray crystallography and increases the efficiency of protein-crystallography beamlines at synchrotron sources like the ALS," explains James Holton of Berkeley Lab's Physical Biosciences Division, who mainly developed Elves while still a graduate student at the University of California at Berkeley. "This in turn should facilitate projects in structural genomics and biology." Holton is in charge of operations at ALS Beamline 8.3.1, an experimental facility powered by hard x-rays ranging in energy from 2.4 to 15 keV (thousand electron volts) from a superbend magnet. This beamline uses an x-ray diffraction technique called "multiple-wavelength anomalous diffraction," which is ideal for imaging proteins and other biological molecules. How to picture a protein With the deciphering of the human genome, as well as the genomes of various other organisms, the next big step in biology is to understand the molecular and cellular functions of the proteins the genes code for. Proteins are the biological molecules that are the building blocks of living cells and serve as the machines that control the chemical processes that make those cells work. To perform their many different functions they will often fold, twist, and corkscrew themselves into a gallery of odd-looking structures that would make M.C. Escher proud. Since form and function go hand in hand for proteins, to understand a protein's function you have to determine its three-dimensional structure. There are several techniques for doing this, but the workhorse method today is x-ray crystallography, in which a beam of x-rays is sent through a crystallized protein. Atoms in the crystal cause the x-rays to scatter, creating a diffraction pattern of dots whose image can be translated by computer into a 3-D model of the protein. When x-ray crystallography was done with low intensity x-ray beams from laboratory x-ray tubes, collecting complete diffraction data sets for a single protein crystal could take months or even years. All this changed with the arrival of synchrotron radiation sources like the ALS, which can generate x-ray beams on the order of a hundred million times more intense than the light from the most powerful x-ray tubes. At that intensity, enough x-ray diffraction data for imaging a typical protein can be collected within an hour, and, for some proteins, within five minutes. This data, however, must then be translated into a 3-D model, a process that, until Elves, has been time-consuming and labor-intensive. The birth of Elves "When I came to Berkeley in 1996 and began doing protein crystallography, I found that the computing side of determining a structure was rate-limiting so I started creating little tools to make mundane tasks go faster," says Holton, a structural biologist who taught himself to become a software programmer. "The tools I created built upon one another, until I finally had a single master program that covered the whole thing."
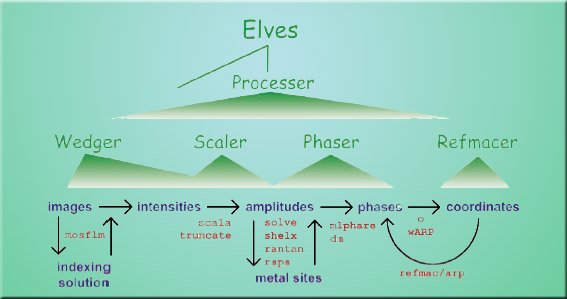
Elves utilizes a three-level hierarchy of programs that can write and optimize scripts for standard crystallography software. The first level consists of four programs called Wedger, Scaler, Phaser, and Refmacer, which handle the four major stages of translating diffraction data into a 3-D structural images. The second level consists of one program, called Processer [sic], which coordinates the work of the first level programs. Elves's main program sits atop the lower levels and runs Processer in an automated mode. "Elves covers a lot of ground in a short time," says Holton. "Its programs systematically explore different crystal symmetry, solvent content, and other critical program parameters. The correct choice is much more obvious this way, so the automated choices made by Elves are usually correct. I don't know of any other crystallographic programs that search as big a parameter space and do it automatically." As a result, Elves has proven more reliable than any previous crystallographic software system—and much faster. A good set of diffraction data can yield a high-resolution 3-D structural image of protein in less than 20 minutes. The emphasis is on good data because without quality diffraction data, Elves cannot produce a quality model. However, Elves can help identify data that is "hopeless" more quickly, which means researchers waste less time working with poor data and more time obtaining good data.
Learning about Elves In addition to being faster and more reliable, Elves is also much easier to learn than other crystallography programs. Holton says the learning curve for software has become "the single greatest bottleneck" in protein crystallography. One of his goals was to minimize the time required to install Elves and get it up and running. This he accomplished through the development of an easy way for users to interact with Elves. Instead of employing a graphical user interface (GUI), as is typical for crystallography software, Holton opted for a conversational user interface (CUI) that enables users to issue software commands in simple English, for example: "Elves, process this data and save to my folder." The CUI also helps make Elves extremely robust. Holton explains that "a GUI can only carry out tasks that its creator had the presence of mind to define a button for. The CUI allows arbitrary complex information to be input into the program without any learning curves." More than 100 scientists are now using Elves, and the system has solved more than 100 protein structures that Holton knows about. Two companies have licensed Elves for commercial x-ray diffraction data analysis. With the structures of perhaps as many as a trillion different kinds of proteins still to be determined, Elves has its work cut out. Already Holton is at work on a system upgrade that will be even easier to learn. Additional information
|
||||||||||||||||||||
| Top | ||||||||||||||||||||