| January, 2007 | science@berkeley lab | | lab a-z index | lab home |
|
|||
| Decoding Breast Cancer Genomes | ||||||||||||||||||||||||||||||
| Contact: Paul Preuss, paul_preuss@lbl.gov | ||||||||||||||||||||||||||||||
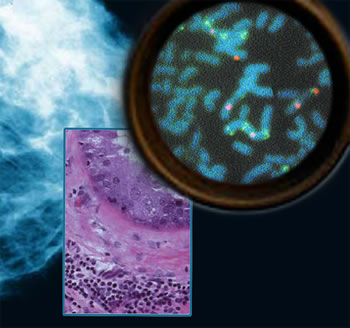
As normal cells turn cancerous and develop into tumors, their genomes accumulate tell-tale changes. Some changes involve gene amplification, meaning they accumulate multiple copies and cause genes to overexpress the proteins they code for. Other changes involve gene deletion, which leads to reduced gene expression. Many of these changes may affect genes that normally regulate cell survival and cell proliferation, processes that, uncontrolled, are hallmarks of cancer.
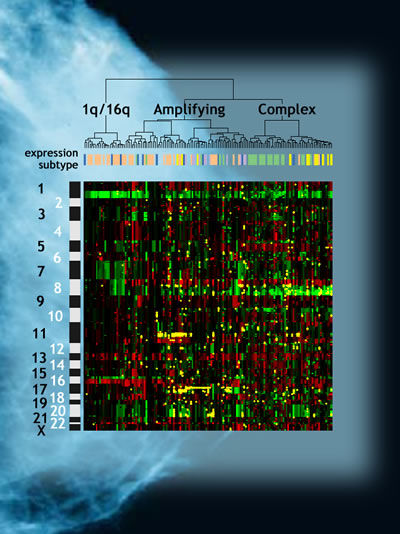
These genomic aberrations are complex but not random. By studying their patterns, researchers are uncovering clues to the many genes involved in breast cancer, how different kinds of breast cancer may progress, and which treatments may be most effective — including targets for promising new drugs. Scientists at Berkeley Lab and the University of California at San Francisco, with colleagues at other institutions, have now linked genomic aberrations in breast cancers to likely clinical outcomes, identified new genes involved in breast cancer, and specified new targets for therapy. In closely related research, they have demonstrated that many of the revealing aberrant patterns found in tumor genomes are well modeled by breast-cancer cell lines. "Our goal is to understand how these abnormalities occur and how the genes associated with them function in breast cancer, so we can identify molecular differences among clinically similar tumors and design effective therapeutic agents against them," says Joe W. Gray, who is director of Berkeley Lab's Life Sciences Division, Associate Laboratory Director for Life and Environmental sciences, and co-leader of the Breast Oncology Program at the UCSF Comprehensive Cancer Center. The new breast cancer genome results were developed in Gray's laboratory. How genome aberrations reflect cancer pathophysiologyThe UCSF Comprehensive Cancer Center's Koei Chin, a member of the Gray lab who is a guest in Berkeley Lab's Life Sciences Division, has conducted research in concert with a score of colleagues from groups at UCSF, Berkeley Lab, the California Pacific Medical Center, and Affymetrix, Inc. Led by Chin, these researchers have shown that analyzing a tumor's genome copy numbers and gene expression can help predict how well a patient’s tumor will respond to current aggressive treatments; the same factors can identify specific genes that can be attacked to improve therapy in patients whose tumors do not respond well to current treatment methods. "Both high-level amplification of some genes — meaning multiple copies of them — and loss of other genes contribute to how breast cancers develop and progress," says Chin. "In some types of breast cancer that don't respond well to current treatments, our research pinpoints genes overexpressed by high amplification in specific regions of the tumor genome, which are potential targets for new or different drug therapies." Chin says that although analysis of gene expression is a powerful method of subtyping breast cancers these days — assays can look at the expression of up to 20,000 genes on a single microassay chip — the power of these gene-based assays to predict treatment outcomes is still not as great as one might hope. "By concentrating on combined analysis of genome copy number and expression, we have been able to identify a small number of genes that improve our ability to better predict clinical outcomes for some cancer subtypes."
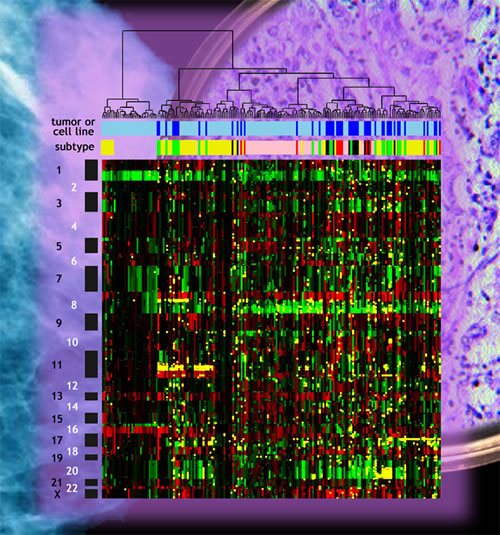
Chin and his colleagues applied microarray techniques to measure changes in genome copy number and gene expression in tumor genomes. By mapping genome copy number abnormalities in tumors from 145 patients, and gene expression in 130 tumors, they were able to identify regions of amplification and deletion that were associated with good and bad clinical outcome. These results may eventually be developed into clinical assays that will allow patients and their physicians to make informed decisions about treatment options. Surgery and radiation have long been the standard treatments for breast cancer; depending on the stage, chemotherapy may be aggressively applied in addition. Anticancer drugs are a significant burden for post-surgery patients, however. Ideally, only the most appropriate therapeutic agents would be selected to treat individual tumors, based on their molecular signatures. Chin and his colleagues found that as the number of genome abnormalities in a breast cancer tumor increases, so does the likelihood of poor treatment outcome. In some tumor subtypes, high-level gene amplification was strongly associated with reduced survival. While patterns of gene amplification and expression correlate well with the known tumor subtypes, the new and more detailed analysis of changes in gene amplification and expression produce more accurate predictions of likely clinical outcomes, by focusing on specific sites and specific genes. In four high-level amplification regions on chromosomes 8, 11, 17, and 20 (namely the regions designated 8p11-12, 11q13-14, 17q11-12, and 20q13), the researchers found 66 genes whose expression had been deregulated. These 66 genes appear to play important functional roles in breast cancer. Although patients with the types of tumors in which there is amplification of these sites generally do not fare well under present therapies, at least nine of the newly identified genes in these regions offer excellent "druggable" targets for new breast cancer therapies. A cell-line systemResearch led by Richard Neve of the Life Sciences Division, in collaboration with colleagues from Berkeley Lab, UCSF, the University of California at Berkeley, the Wayne State University School of Medicine, the University of Texas Southwestern Medical Center, the Georgetown University School of Medicine, and the University of Michigan Medical School, has established that genome analyses based on a library of 51 breast cancer cell lines maintained in vitro are substantially similar to analyses based on tumor tissues. Thus, information gained from cell-line experiments can greatly complement the analysis of a primary tumor and improve clinical decisions. "By using data from a growing library of cell lines, we can better understand how specific genomic or gene expression abnormalities influence cancer behavior," says Neve. "This can lead to a better treatment plan and a better understanding of possible outcomes, resulting in a better choice of therapeutic drugs."
Because the cell lines retain most of the recurrent patterns of genomic changes present in primary tumors, says Neve, "The genomic characteristics of the cell lines reflect those of the tumors — not perfectly, but quite well, so we can use these to investigate how these characteristics influence the cancer biology." Oncogenes associated with the development of cancer are among those amplified in the genomes of cancerous cells, while tumor suppressors are among those deleted or suppressed, Neve says. "We come out with a long list of potential oncogenes and tumor suppressors — a list that identifies many genes not previously thought to be associated with breast cancer." Not only does this information suggest novel drug targets, it can improve patient selection for clinical trials of a range of new drugs. "We can pull out a gene profile and find the patients most likely to respond, which could save money and could also get much better results for those patients." Neve says, "Research using cell lines has suffered from poorly maintained and annotated stocks. We have developed a well characterized reference stock of cell lines, and we've found that the best way to track and identify a particular cell line is to look at its DNA fingerprint. We have also amassed a large body of biological information for these cell lines. The more people who share this data, the better, so we are putting all our information on the web." Focusing on gene amplification and gene expression in both primary tumors and in a wide range of existing cell lines opens a new path for studying how breast cancers develop, for predicting the outcomes of breast cancer treatments, for suggesting promising new targets of drug therapy, and for honing clinical trials for more meaningful results. Koei Chin and his colleagues were supported in this work by the National Institutes of Health, the Department of Energy's Office of Science, and the Avon Foundation. Research by Neve and his colleagues was supported by DOE's Office of Science and the National Institutes of Health. Additional information
|
||||||||||||||||||||||||||||||
| Top | ||||||||||||||||||||||||||||||