| July 26, 2006 | science@berkeley lab | | lab a-z index | lab home |
|
|||
| Unraveling the Complex Codes of Human Gene Expression | |||||||||||||||||||||||||||||||||||
| Contact: Paul Preuss, paul_preuss@lbl.gov | |||||||||||||||||||||||||||||||||||
Sequencing the human genome is a major challenge; although a first draft was published in 2001, the details are continually being refined. Quite another challenge is to find all the human genes, about 30,000 in number; that process too is ongoing. Much more complex than either of these tasks, however, is to understand how human genes are regulated.
Debopriya Das, a member of Berkeley Lab's Life Sciences Division, recently published a new computational approach to human gene regulation that he developed with Michael Zhang of Cold Spring Harbor Laboratory and Zaher Nahlé of Washington University at St. Louis, when Das was a postdoctoral fellow in Zhang's lab. At Berkeley Lab, Das is using the new approach to study the regulation of genes in breast cancer. Humans are different from yeast"Gene regulation is a far more intricate process in humans than in simpler organisms like yeast," says Das. "With lower eukaryotes, if you have the genome sequence, you can integrate this data with global expression profiles and use one of several computational approaches that have already been developed to decipher the cis-regulatory elements with reasonable accuracy." Cis-regulatory elements are short elements on the DNA, usually upstream of the gene; they make up part of the gene's promoter, its "on/off" switch. Genes are expressed when they are transcribed into messenger RNA, which is subsequently read by the cell's machinery to make proteins. (To prevent the expression of an unnecessary or harmful gene the cell may choose to actively downregulate it, deliberately flipping it into the off state.) With a simple organism like yeast, there are multiple instances where a single transcription factor (itself a protein) is enough to turn on the gene by binding to the promoter's cis-regulatory elements. A general term for these transcription-factor binding sites is "motifs," short sequences of bases. "A motif is like a word, written by the four letters of DNA, the bases," says Das. "In lower organisms, the spelling of that word is mostly fixed." If different genes have the same motif, the same transcription factor can turn on any of them. Das and his colleagues call the elements that work together to control gene expression "transcriptional subnetworks." Transcriptional subnetworks are comprised of motifs, the transcription factors that bind to those motifs, and the genes (and processes) that they directly regulate. (These genes are often called the direct targets of the transcription factors.) In mammals, especially in humans, identifying transcriptional subnetworks is no easy task. What makes human gene regulation hard to decode?"One aspect is that in humans the motifs are significantly 'degenerate,'" says Das. "That means the cis-elements in the promoter regions of different genes that are recognized by the same transcription factor have a lot of substitutions in their sequences." A mammalian motif typically has five to 15 base pairs relevant to binding — five to 15 key letters in the motif word. Degenerate motifs are words that are spelled differently but have the same "meaning" — they bind to the same transcription factor.
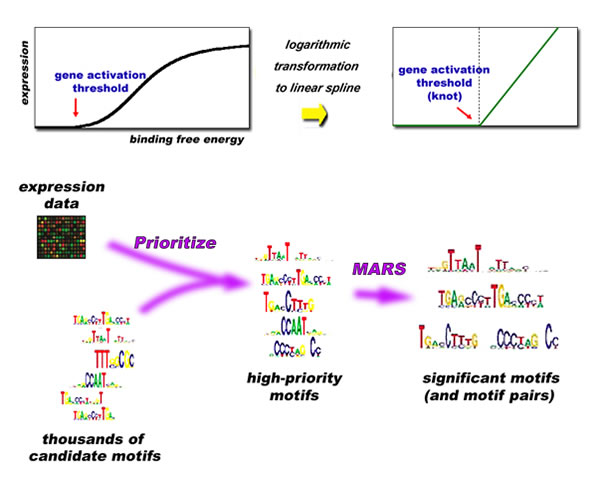
How strongly a transcription factor binds to a motif is known as its binding free energy. Binding free energy may be high when a transcription factor fits a specific sequence of bases — a word spelled in a certain way — but as motifs become degenerate and relevant letters are substituted (adenine for thymine, for example, or cytosine for guanine) binding energy decreases, and less of the transcription factor binds successfully. In mammalian gene regulation, interactions among the transcription factors themselves are also critical. "It usually takes two or more transcription factors working together, each with its own binding sites in the promoter, to cause a gene to express," says Das. Finally, he says, "the multicellular architecture of mammals complicates the problem. Most cells in the human body carry the entire genome, all 30,000 genes, but not all these are expressed." Thus there are regulatory subnetworks specific to different cell types. To add to the complexity, regulatory cross-talk across different tissues is often essential for generating a specific physiological response. Tackling complexity with MARSMotif-MThe algorithm developed by Zhang, Das, and Nahlé, which they call MARSMotif-M, overcomes the difficulties of decoding human gene regulation in a series of steps, using a mathematical technique known as Multivariate Adaptive Regression Splines (MARS). A linear spline is a mathematical function that can represent the interactions of complex physical phenomena. It has zero function before (or after) an on-off point called a "knot," which is labeled with the knot-like Greek letter xi, ξ, and a straight-line change in magnitude after (or before) the knot. "In other words," says Das, "splines provide a convenient functional representation of an on/off switch, especially when the output of the switch changes almost linearly with the inputs — in our case, binding free energies — in its on state."
Because splines can model the sum of the activity of several variables, they are well suited to represent the interactions of multiple transcription factors in terms of their DNA binding motifs, including degenerate motifs. No prior knowledge of how genes are regulated in the specific cell type that is being interrogated is necessary. All that is needed is a library of promoter sequences of genes. Das and his colleagues tested MARSMotif-M on two distinctly separate cases of gene expression in humans, one tissue-specific, the other looking at different stages of the human cell cycle. The tissue-specific case focused on normal adult liver cells, based on gene expression data from a DNA microarray "snapshot." In the microarray system, expression levels are gauged by measuring the amounts of messenger RNA associated with genes in different portions of transcribable DNA. In the case of liverThe researchers employed a set of 521 candidate motifs previously identified as having a significant role in gene expression (in all vertebrates, under a wide variety of conditions). To find which of the candidate motifs were likely to be actively bound, the motifs were first matched to promoter sequences from genes selected from the most active thousand genes in 79 different human tissues. Next the motifs were scored by fitting linear splines to each to see how well each motif by itself could explain the variation in expression levels across genes observed in the DNA microarray. Remarkably, the candidates fell into two quite separate groups, with a gap between the high-scoring group and all the others. Searching for optimal spline models among the high-scoring motifs, the researchers identified three single motifs and five pairs of motifs that stood out as good predictors of gene expression in liver. The motifs corresponded to specific transcription factors. In this "blind" search, not guided by any prior knowledge of liver genes or motifs, some of the motifs identified had in fact been earlier characterized as involved in liver regulation. But two of the motif pairs had never previously been associated with gene expression in liver. The next step was to identify the target genes of these transcription factors. "This can be done experimentally, but it's tedious, especially when dealing with degenerate motifs. Our algorithm makes it much simpler to distinguish a transcription factor's true targets from possible false ones," says Das.
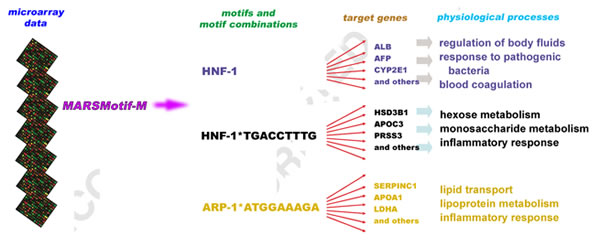
In the case of liver, the researchers first looked at a transcription factor called HNF-1 (for hepatic nuclear factor, which signifies association with DNA in the nuclei of liver cells). They uncovered 29 target genes for HNF-1, 20 of which had been characterized before — the remaining nine, however, were previously unsuspected. Employing an additional 1,440 candidate motifs generated from computer programs, Das and his colleagues identified two novel motif pairs that were located in the promoters of genes that regulate such vital liver processes as sugar metabolism, lipid transport, and response to inflammation. This discovery could shed light on the transcriptional basis of cell toxicity and inflammation in certain unusual liver conditions, and thus help in designing better therapeutics for the treatment of pathological conditions like diabetes, insulin resistance, and obesity. On to the cell cycleIn a second test case, the researchers looked at gene expression during different stages of the cell cycle of cultured HeLa cells (cervical cancer cells used in many cancer biology research projects). They applied MARSMotif-M separately to each of the 19 different time points of the cell cycle to discover motifs and combinations of motifs active at that specific point in time. In this way they identified a large number of individual motifs and motif pairs, some of which operate almost all the way through the cycle while others come into play in different combinations at different stages. They also identified the target genes associated with these motifs and pairs. "Target genes for a particular transcription factor can be different at different stages in the cell cycle, because the same factor can partner with different transcription factors in response to different cellular contexts," says Das. "Moreover, at different stages the binding-energy threshold for gene activation" — the knot on the spline — "changes as well." MARSMotif-M can conveniently model such condition-dependent gene regulation. For example, the researchers were able to independently confirm the role of the E2F family of transcription factors at several distinct phases of the cell cycle. And along with that, they identified previously unsuspected direct targets of E2F. Two of the new targets, which they later validated experimentally using biochemical assays, are genes known to play a role in the progression of liver cancer. This added knowledge may help to design new diagnostics or therapies for liver cancers. A new tool for discoveryIn both the tissue-specific liver case and the cell-cycle case, the motifs and motif pairs discovered by the new algorithm were found de novo, starting from scratch, using candidate motifs from data sets derived from many different tissues — and even generated by computer programs.
"We blinded ourselves when we applied the algorithm to the expression data," Das explains. "In some cases what we found was validated by previous work, and in other cases we made new discoveries, which we were able to validate with our own experiments." Says Das, "The new algorithm provides a way to overcome the critical difficulties presented by degenerate motifs in mammalian cells, and by the interaction of transcription factors essential for activating a mammalian gene." But many challenges remain, he says, naming a long list of other factors that make human gene expression so complex:
"And there are others," says Das. "But given that in mammals, most motifs haven't even been discovered yet, MARSMotif-M is an important first step in dissecting the complex processes of human gene regulation." Additional information
|
|||||||||||||||||||||||||||||||||||
| Top | |||||||||||||||||||||||||||||||||||