| lab a-z index | phone book |
| December 20, 2004 | news releases | receive our news releases by email | science@berkeley lab |
|
|||
|
|||
| Solving the Mechanism of Rett
Syndrome How the First Identified Epigenetic Disease Turns on the Genes That Produce its Symptoms |
|||||||||||||||||||||||||||||||||||
| Contact: Paul Preuss, (510) 486-6249, paul_preuss@lbl.gov | |||||||||||||||||||||||||||||||||||
|
BERKELEY, CA – Sometime between the age of 6 and 18 months, after a period of seemingly normal development, girls affected with Rett Syndrome lose interest in play; they gradually become withdrawn and anxious, develop autistic-like behaviors, and acquire specific symptoms like repetitive teeth-grinding and hand-wringing. This devastating neurological disease affects one in 15,000 female children.
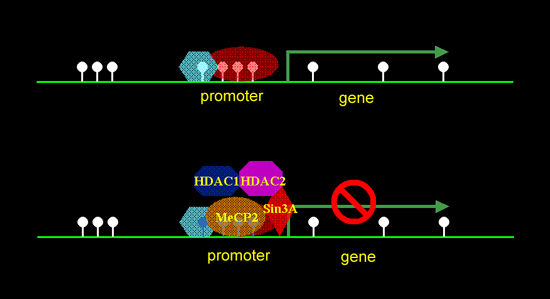
Just five years ago, Rett Syndrome was tracked to mutations in a gene on the X chromosome, MECP2. But how this gene, not previously associated with the brain or nervous system, could cause a neurological developmental disorder remained a puzzle. Now, a team of scientists with the U.S. Department of Energy's Lawrence Berkeley National Laboratory has developed new methods and overturned mistaken assumptions to discover how the product of this gene, the protein MeCP2, can remodel chromatin, the material that makes up chromosomes. For the first time a human disease — Rett Syndrome, the first identified epigenetic disease — has been linked to specific defects in the three-dimensional folding of chromatin. The research was supervised by Terumi Kohwi-Shigematsu, a biochemist with Berkeley Lab's Life Sciences Division; it reveals how mutated MeCP2 protein represses genes, and identifies some of the most important of those genes. Kohwi-Shigematsu and her colleagues, Shin-ichi Horike, Shutao Cai, Masaru Miyano, and Jan-Fang Chen, report their results in the January issue of Nature Genetics (.pdf). How MECP2 worksSo-called CpG "islands" are found at the promoter regions of many housekeeping genes, which code for proteins essential to cell function. They contain high densities of cytosine and guanine base pairs, called CpG dinucleotides. MECP2 stands for "methyl CpG-binding protein 2;" as its name indicates, it can bind to these base pairs when methyl groups (CH3) are attached to them. Normally, CpG dinucleotides in CpG islands are not methylated and the genes are active. If CpG islands are methylated, however, they attract MeCP2 proteins, which bind additional proteins that repress gene transcription and turn the promoter off. Thus MeC2P is thought to be a key player in assembling the protein factors that silence transcription. Because MeCP2 binds to methylated CpG dinucleotides, its effects are not dependent on the primary sequence of DNA. "One proposal for how defective or absent MeCP2 protein might cause Rett Syndrome was that, by failing to attach to methylated CpG dinucleotides, it would fail to repress inappropriate gene expression in the brain," says Kohwi-Shigematsu.
For some genes, called imprinted genes, their expression status depends on whether the gene came from the maternal or paternal allele, with the two forms often having differently methylated CpG islands at or near their promoters. A leading hypothesis of how mutated MECP2 could produce Rett Syndrome is that the mutation disrupts this imprinting mechanism. An imprinted gene, one with a methylated promoter, is usually silent. If defective or missing MeCP2 protein were to fail to silence an imprinted allele, the expression of the gene would double. Failure to repress imprinted alleles has been implicated in several neurological disorders. "MeCP2, the protein coded for by the MECP2 gene, is expressed in many tissues, including brains," says Kohwi-Shigematsu. "People thought it was a general repressor that regulates gene expression throughout the body. Yet the main syndrome of Rett patients pointed to neurodevelopmental problems after birth. So our first challenge was to find out which genes MeCP2 directly regulates in the brain, and how it regulates them." Searching for targetsThe researchers examined hundreds of MeCP2 binding sequences in the brains of mice. In wild-type mouse brains they found that the MeCP2 protein binds in the vicinity of some five dozen genes, several of which reside in a cluster of imprinted genes on mouse chromosome 6 (corresponding to a region of human chromosome 7). When the binding sites in wild-type mouse brains were compared to the same sites in MeCP2-null mice — "knockout" mice bred with no MecP2 gene and thus no MeCP2 protein — one region in particular stood out: expression of the adjacent genes Dlx5 and Dlx6 almost doubled in the knockout mice. The identification of the Dlx5 gene in mice was highly suggestive, since in humans the DLX5 protein plays an important role in the synthesis of GABA, gamma-aminobutyric acid, an important neurotransmitter.
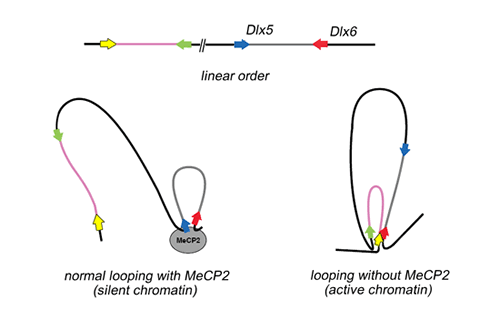
The researchers sought similar effects in cells from Rett Syndrome patients, substituting cultured lymphoblasts (immune-system cells) for inaccessible brain cells. In humans, normally only the maternal allele of DLX5 expresses, because the paternal allele is imprinted. But lymphoblasts from many Rett Syndrome patients exhibited a much higher rate of DLX5 expression. When an imprinted gene nevertheless continues to express, the phenomenon is called "loss of imprinting." The researchers had now identified at least one gene targeted by MeCP2 where, if the protein were missing or defective, the result might lead to misregulation in the production of the neurotransmitter GABA. But the mechanism by which normal MeCP2 acts to regulate the DLX5 gene and how this regulation goes awry were still to be determined. One thing was clear: MeCP2's propensity to bind to methylated CpG islands played no part. CpG islands near Dlx5 and Dlx6 were found — quite unexpectedly — to be completely unmethylated in wild-type mice, knockout mice, and the human lymphoblast cell line. Even where individual CpG base pairs in the region outside CpG islands were methylated, there were no differences in methylation patterns between the maternal and paternal alleles. Into the third dimensionMethylation in CpG dinucleotides was clearly insufficient to explain how MeCP2 normally regulates the DLX5 gene. The researchers pursued other possibilities. To do the actual work of gene suppression, MeCP2 acts in concert with a histone deacetylase protein, HDAC1, and other proteins. Histones are proteins in chromatin equipped with little "tails" that, when attached to an acetyl group (CH3OH), relax to allow the chromatin structure to become open or less compacted. When acetyl groups are not present, the deacetylated chromatin condenses. Genes are not expressed in silent chromatin because their DNA is tightly constrained by deacetylated histones, which prevent transcription enzymes from accessing the gene. Kohwi-Shigematsu and her team found a discrete deacetylated region of chromatin in the Dlx5/Dlx6 gene neighborhood that coincided with the main MeCP2 binding site. In MeCP2-knockout mouse brain, this silent chromatin was missing. Kohwi-Shigematsu and her colleagues decided to investigate what effect MeCP2 might have on the structure of chromatin near this site in three dimensions. To this end, they created a complex new assay, the chromatin immunoprecipitation-combined loop assay, and used it to uncover a remarkable arrangement of chromatin loops in the Dlx5/Dlx6 neighborhood.
In wild-type mice, they found that MeCP2 is required for the formation of a loop of silent chromatin between the Dlx5 and Dlx6 genes, by bringing together two sequences separated by more than 10,000 base pairs. This silent chromatin loop configuration could not be formed in MeCP2-null brain, and in MeCP2-null mice, the two genes are highly expressed. Mechanisms of Rett SyndromeRett Syndrome symptoms can be associated with the failure of mutated MECP2 to regulate transcription of a specific gene, DLX5, one allele of which is normally imprinted. Without the MeCP2 protein, production of the Dlx5 protein is increased, which must influence production of the neurotransmitter GABA and may also affect the expression of other, related genes in the DLX family with consequences for the development of the brain. Absent or defective MeCP2 allows increased expression of the DLX5 gene through the loss of a loop of silent chromatin, and the activation of additional neighboring chromatin. This dramatic rearrangement in the DLX5/6 gene neighborhood may also involve other disturbances in gene expression. Similar rearrangements, with equally profound effects, may be found among the five dozen other genes targeted in the brain by MeCP2, which are now under active investigation by Kohwi-Shigematsu and her colleagues. "Loss of silent chromatin looping and impaired imprinting of DLX5 in Rett syndrome," by Shin-ichi Horike, Shutao Cai, Masaru Miyano, Jan-Fang Chen, and Terumi Kohwi-Shigematsu, appears in the January issue of Nature Genetics (.pdf). Berkeley Lab is a U.S. Department of Energy national laboratory located in Berkeley, California. It conducts unclassified scientific research and is managed by the University of California. Visit our website at http://www.lbl.gov. |
|||||||||||||||||||||||||||||||||||
| Top | |||||||||||||||||||||||||||||||||||