|
February 28, 2003
|
||
|
 |
 |
| "ProteinShop": solving protein structures from scratch | ||
| Contact: Paul Preuss, paul_preuss@lbl.gov | ||
|
|
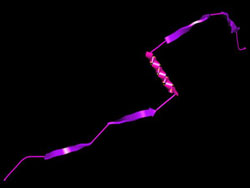
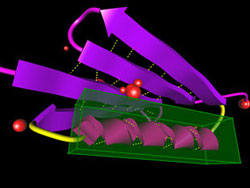
ProteinShop, a computer visualization tool for manipulating protein structures, is closing in on one of biology's cherished goals: completely determining an unknown protein's shape from its gene sequence. Silvia Crivelli of the Visualization Group in Berkeley Lab's Computational Research Division says a major step forward came when "we copied concepts from robotics. When you move a robot's arm, you move all the joints, like your real arm." After a year of work on ProteinShop, Crivelli says, "we were able to apply the same mathematical techniques to protein structures."
It's just one of the components the ProteinShop vehicle uses to help competitors in the hottest race in biological computation — the "Grand Prix of bioinformatics" known as CASP, the Critical Assessment of Structure Prediction. In 1994, the challenge of predicting finished protein structures from gene sequences gave rise to the unique CASP scientific competition. Teams of biologists and computer scientists from around the world try to beat one another to the fastest and most accurate predictions of the structures of recently found but as-yet unpublished proteins. The number of competitors has more than quintupled, from fewer than three dozen groups in 1994 to 187 in last year's CASP5. At CASP5, a team led by Teresa Head-Gordon of Berkeley Lab's Physical Biosciences Division, who is also a professor of bioengineering at the University of California at Berkeley, employed the Global Optimization Method developed by Head-Gordon and her colleagues, including Crivelli from Berkeley Lab and Bobby Schnabel, Richard Byrd, and Betty Eskow from the University of Colorado. Instead of depending heavily on knowledge of protein "folds" (three-dimensional sets of structures) already stored in the Protein Data Bank (PDB), the Global Optimization Method uses initial protein configurations provided by ProteinShop, which was created by Wes Bethel and Crivelli of Berkeley Lab along with Nelson Max of Lawrence Livermore National Laboratory and the University of California at Davis, and UC Davis's Bernd Hamann and Oliver Kreylos. With a ProteinShop jumpstart, Head-Gordon's CASP5 team was able to predict the configurations of 20 new or difficult protein folds ranging from 53 to 417 amino acids in length. The team ranked 15th in the competition overall, which, says Crivelli, "is great for a method that doesn't use much knowledge from the PDB." Structure by the numbers A protein's shape is what determines its function. Yet a one-dimensional string of amino acid residues, as specified by a gene's coding sequences, does not on the face of it reveal much about three-dimensional shapes.
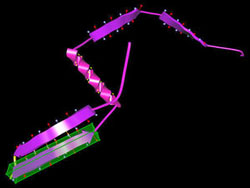
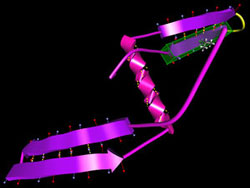
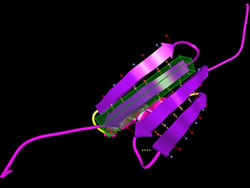
Swimming in a watery environment, the amino-acid string — the protein's primary structure — quickly twists into familiar shapes known as alpha helices and beta sheets, striving to reduce attractive and repulsive forces among the linked amino acids. The result is local regions where the energy required to maintain the structure is minimized. The secondary structures swiftly fold up into a three-dimensional tertiary structure; for most proteins this represents the "global" minimum energy for the structure as a whole. (Some complicated proteins like hemoglobin, assembled from distinct components, have a quaternary structure as well.) By studying thousands of known proteins, biologists have learned to recognize sequences of amino acids that are likely to form alpha helices, plus those that form the flat strands that arrange themselves side by side in beta sheets. This secondary-structure information can be stored in data banks and applied to unknown protein folds. But the so-called coil regions that link secondary structures in an unknown protein are not so easily predicted. The more amino acids in the protein, the more local energy minima there are, and the harder it becomes to calculate the global energy minimum. The global optimization method tackles the problem in two distinct phases. During the setup phase, the program generates likely secondary structures from a given sequence of amino acids, which are combined into several approximate configurations.
During the next phase, the overall energy of the structure is reduced step by step. Global optimizations are performed on subsets of the angles among the amino acid residues (dihedral angles) which are predicted to make up coil regions. Eventually, because the energy function is not perfect, several tertiary structures are proposed. Before ProteinShop, the set-up phase of the global optimization method was immensely time-consuming. Because a protein's primary structure contains dozens or hundreds of acid residues, thus thousands of atoms, the initial phase required computer runs of hours or days before settling on secondary structures. Moreover, while the prediction of alpha helices was straightforward, beta sheets were hard to assemble from beta strands, and their configurations were less certain. In CASP4, in 2000, the team predicted the structures of only 8 folds, the longest containing 240 amino acids. Spaghetti at last The Visualization Group developed ProteinShop in preparation for CASP5. ProteinShop incorporates the mathematical technique called inverse kinematics — well known not only in robotics but also in video gaming. Inverse kinematics is used to predict the overall
movements of structures consisting of jointed segments, for example the
fingers, arms, and shoulders of a robot or an animated figure. By taking
into account the degrees of freedom permissible at each joint, contortions
that don't break limbs or penetrate bodies can be predicted.
"The difference is that with a robot you have maybe 10 or 20 joints, but in a coil we often have long regions, 80 amino acids," Crivelli says, "and we want all of the dihedral angles among them to move in a concerted way." In ProteinShop, the secondary structures and coils are built up by adding amino acids to the structure one at a time, treating each as a jointed segment of a flexible structure. Within seconds a "geometry generator" module incorporates predicted secondary structures, or fragments of them, in the string. "The whole thing looks like you could just move it around like spaghetti," says Crivelli. "But before we incorporated reverse kinematics, if you tried to move a protein configuration, it broke." Now the process works fast enough to be truly interactive, allowing the user to alter the dihedral angles between individual amino acids. In difficult cases like the assembly of beta strands into sheets, the user can manipulate the conformation to achieve the best, least energetic fit. Moreover, the user can play with the entire "preconfiguration," dragging whole secondary assemblies into new relationships without breaking previous structures. "Once we have the alpha and beta structures,
we want to leave them alone," Crivelli says. "Mainly we work
on the coil regions."
The ProteinShop program allows for various styles of illustrating the fold and its parts, and it helps out the user by sending plenty of signals. In one mode, little orange spheres pop up when the user has forced atoms to collide: the more energy-expensive the collision, the bigger the orange sphere. Manipulation can also be simplified by hand-selecting residues for automatic bonding. The future of global optimization The second phase of the global optimization method — seeking the global energy minimum — remains a computational challenge. The basic approach to finding the optimal energy for a tertiary structure is to repeatedly tweak the energy budgets of many smaller regions, incorporating each improvement until there is no further change. This "physics-based" method depends only on energy calculations, not on knowledge of existing folds. In practice, to do such calculations for every part of a structure would take much too long. The sophisticated procedures developed by Head-Gordon's team optimize only randomly sampled, small regions. Thus it's not certain that the end result is the true global optimum. Even so, global optimization of an unknown protein of moderate size may require weeks of computer time. "We are working out new methodology that will
combine our physics-based approach with knowledge-based methods,"
says Crivelli. "By recognizing structures and fragments that are
known to work, we won't have to calculate every angle from scratch. The
tool will be highly interactive, displaying energies and saving minima
as the user finds them. It will be organized as a guided search through
a dynamically evolving tree, basing new structures on previous ones that
have been shown to work for the fold."
To enable this high degree of interaction, still higher performance will be required from the global optimization method's already high-performance parallel-processing codes. The result will have implications for scientific problems far beyond the successful solution of unknown protein structures. Meanwhile, ProteinShop is being tooled up for the challenge of CASP6. Many groups who competed in CASP5 are already interested in what the powerful ProteinShop can contribute to their own methods for running the race to CASP6's protein-prediction finish line. Additional information
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||